Researchers Tackle the Flu with Breakthrough Virus Simulations
Published Date
Article Content
A team of researchers from the University of California San Diego and the University of Pittsburgh offers a new approach for developing treatments for influenza. This version of the flu called pandemic Influenza A H1N1 2009, or pH1N1, differs from the Coronavirus, and has been known to kill hundreds of thousands of people each year. The Center for Disease Control estimates that it affected nearly 61 million people, resulting in more than 12,000 deaths, in the United States alone. The H1N1 virus also circulates among bird and pig populations, causing seasonal epidemic and pandemic outbreaks, such as the 1918 Spanish Flu, which killed millions of people worldwide.
Additionally, the increasingly widespread resistance to antiviral medications is compounding this threat, thus requiring the development of novel approaches for the prevention and treatment of influenza virus infection.
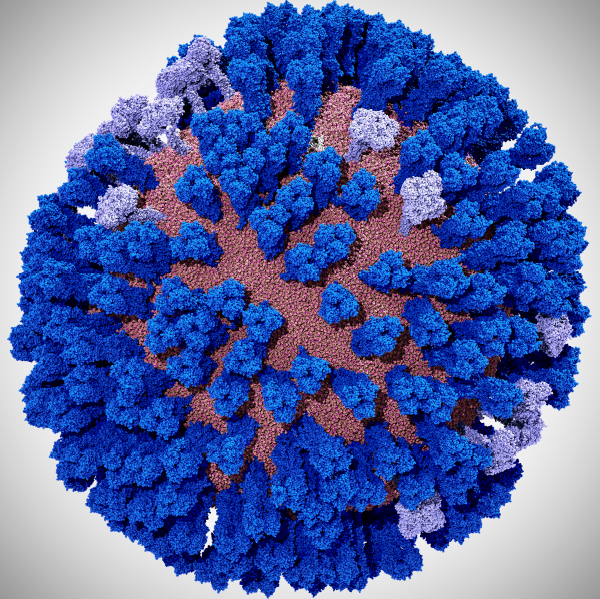
Mesoscale simulations enhance conformational sampling of the viral glycoproteins. This image shows a fully intact all-atom model of the influenza A H1N1 2009 (pH1N1) viral envelope, containing over 160 million atoms, without explicit water molecules. Photo courtesy of Lorenzo Casalino, UC San Diego
The process of creating new therapeutics that effectively fight the flu virus involves scientists experimenting with molecular simulations. In a recent study, led by UC San Diego’s Rommie Amaro, researchers broke new ground with their simulations in terms of size, complexity and methodological analyses of the simulated components. Their findings are published in Central Science, a journal of the American Chemical Society.
Working with pH1N1, the research team examined two specific binding sites among the rich molecular environment of the virus. They better characterized these two sites by developing an all-atom, solvated and experimentally based integrative model of pH1N1.
“Even just building this system from the ground up was quite challenging as it required us to integrate different types of experimental data, at different resolutions,” said Amaro. “This model itself was useful to understand the physical arrangement of molecules in the virus, which had never been done before at this level of detail.”
The professor of chemistry and biochemistry at UC San Diego explained that to aid in drug discovery, researchers often create highly detailed models of proteins and the drugs with which they interact. In most cases, however, to do this, they greatly simplify the system and focus on one single drug target. The work by Amaro and colleagues is considered a breakthrough because it models the entire influenza viral envelope, and its many components, without sacrificing the details of each target. This effort therefore gives new views into complexity of the individual drug targets, within their native environment.
Once the system was built, the researchers employed one of the largest and most powerful supercomputers, the petascale Blue Waters system at the National Center for Supercomputing Applications (NCSA) at the University of Illinois at Urbana-Champaign, to simulate how all the atoms in the viral envelope move.
Amaro noted the novelty of accomplishing the simulation relative to the large scale of the biological system, one of the largest ever to be simulated.
“From a more scientific standpoint, it allowed us to see how the different flu proteins interacted with each other on the viral surface,” said Amaro.
Video simulation related to new Influenza A, pH1N1 research findings by UC San Diego’s Rommie Amaro and her collaborators. Video courtesy of Lorenzo Casalino, UC San Diego
The team also used the many copies of each protein in the virus together with a statistical analysis technique known as Markov state modeling to estimate how quickly different parts of the viral proteins moved.
“This was useful because it allowed us, for the first time, to characterize how quickly one particular loop known as the ‘150-loop’ opened and closed,” said Amaro. “We care about the motion of this loop because the loop is right up next to where drugs, like Tamiflu, bind to deactivate the protein.”
Amaro added that the research also provided fundamental insights into the understanding of substrate recognition processes for this vital influenza drug target, suggesting a new strategy for the development of anti-influenza therapeutics.
“Our study also provides new evidence that an often-overlooked so-called ‘secondary site’ may be the first place the natural substrate of the flu binds. Thus a novel viable therapeutic strategy may be to design molecules that effectively block that site,” said Amaro.
Blue Waters Director William Kramer said that few supercomuters are capable of the petascale performance required for these research results. “We are thrilled Blue Waters, both the computer and the support staff, was able to contribute to this groundbreaking success. To our knowledge, a molecular dynamics simulation on so grand a scale has never been attempted, let alone completed,” he said.
This work was funded in part by the National Institutes of Health Director’s New Innovator Award Program (NIH DP2 OD007237) and the National Biomedical Computation Resource (NIH P41 GM103426). Computing support on the Blue Waters Petascale Computer was provided through the National Science Foundation (OAC- 464 1811685). The researchers recognize the NSF for access to the TACC Stampede supercomputer (CHE060073N) and the Center for Research Computing at the University of Pittsburgh for a computing allocation. The authors also acknowledge Robert Malmstrom for providing sample MSM-building scripts and useful discussions.
The full research team included Amaro; Jacob Durrant, University of Pittsburg’s Department of Biological Sciences and Lorenzo Casalino, Pek Ieong and Abigail Dommer from UC San Diego.
Share This:
You May Also Like



Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.