UC San Diego Chemists Take Aim at Drug Predictions
Targeting computational challenges in drug discovery results in new mode of protein structure analysis
Published Date
Article Content
As many American consumers know, “pharma” means the big business of drug therapy. According to the U.S. National Library of Medicine, drug discovery is no longer a “target-and-mechanism-agnostic approach,” rooted in ethnobotanical information and infused with “serendipity.” Instead, drug discovery has grown into a hypothesis-driven, target-based approach. This, thanks to advances in molecular biology, knowledge of the human genome, data computation and impactful changes in the pharmaceutical industry. Now, according to PharmaExec.com, the “holy grail” of computer-aided drug design (aka CADD) is the accurate prediction of the bond between a drug molecule and its protein target.
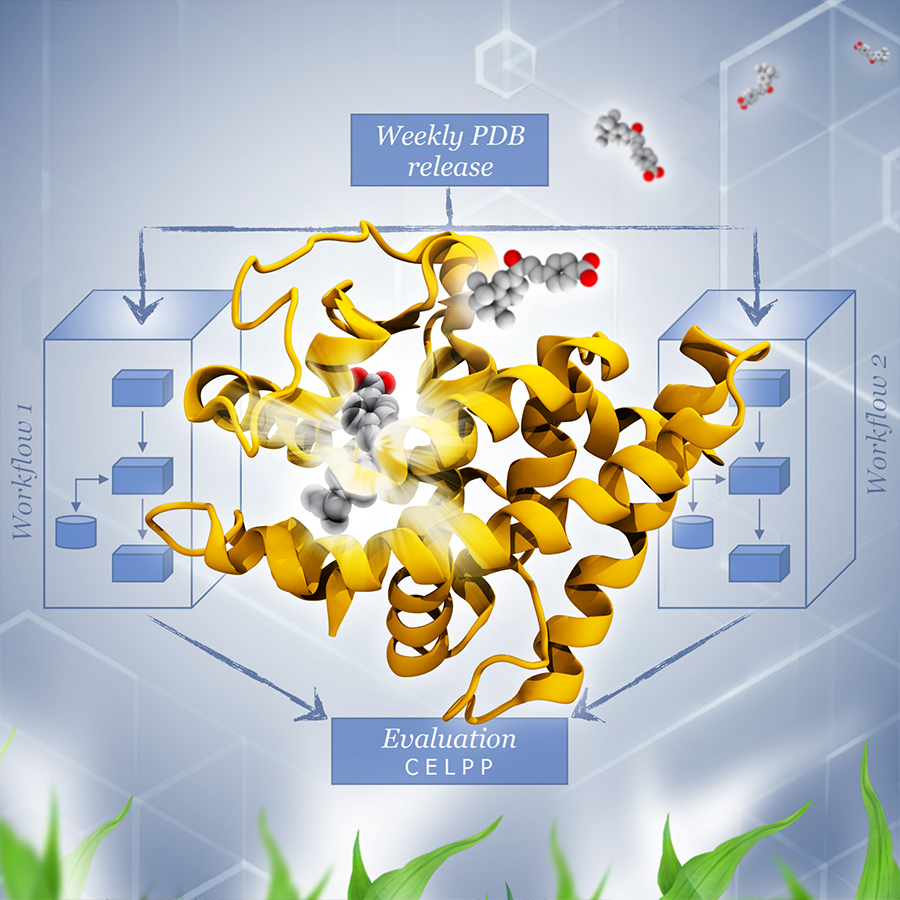
Docking calculations can accelerate drug discovery by predicting the bound poses of ligands for a targeted protein. However, it is not clear which docking methods work best. The Continuous Evaluation of Ligand Protein Predictions (CELPP) takes advantage of the continuous stream of data in the Protein Data Bank (PDB) to host a weekly public challenge designed to address this question, and ultimately, accelerate the creation of new and safer medications. Image by Lorenzo Casalino, UC San Diego
This is where academia comes in—namely Chemists and Professors Rommie Amaro and Michael Gilson at the University of California San Diego. Together with a team of scientists, they contribute to drug discovery by converting their basic science research findings into potential new medical treatments. In fact, Amaro and Gilson’s latest research results, now published in Structure, present promise for addressing the challenges of computational methods in the drug discovery process with a new prospective, or “blinded,” prediction challenge called Continuous Evaluation of Ligand Protein Predictions (CELPP).
This is a method that scientists can use to ease the synthesis and evaluation of the algorithms, chemistry and technology needed to predict the bound poses of ligands—molecules that bind to other molecules—within a targeted protein. This is necessary in order for a new drug to be designed. It’s all very technical, but basically things called docking calculations can speed up drug discovery by predicting the bound poses of ligands. But chemists aren’t sure which docking methods work best. This is where CELPP is already proving to be useful.
“The discovery of a small molecule that binds a disease-related protein with high affinity is a key step in many drug discovery projects,” said Amaro.
The distinguished chemist explained that there are two main components of the computational challenge of structure-based ligand design: 1) the prediction of bound pose of a specific ligand and 2) using the predicted pose to assess the ligand’s binding affinity for the targeted protein. According to Amaro, both components have been intensively researched academically and commercially, with interest growing in automated workflows that prize a particular method.
What’s special about CELPP is that, through a new collaboration with the Research Collaboratory of Structural Bioinformatics (RCSB) Protein Data Bank (PDB), researchers now have early access to soon-to-be-released structures that are released on a weekly basis in the PDB.
“By taking advantage of this already existing flow of structural data into the PDB, we’ve effectively increased—by orders of magnitude—the data that software developers can use to test their algorithms and prized workflows,” noted Gilson. “In doing so, CELPP sets the stage for a technological leap forward in our ability to predict how a drug binds to its target.”
The study led by Amaro and Gilson details the challenges, the automation used to enable smooth predictions, comparative results for various docking workflows, and implications and directions for what they call a true “community science project.”This study was supported by the National Institutes of Health (grant U01 GM111528).
Share This:




Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.