A New Phase
Emerging technologies, social imperatives and the next pandemic are changing how clinical trials look and work.




Published Date
Article Content
During his reign (605 BCE-562 BCE), Nebuchadnezzar II, King of Babylon, Sumer, Akkad and all of the universe, decided that a diet consisting entirely of meat and wine ensured good health, and so ordered his subjects to eat nothing more or less.
Not everyone agreed and Nebuchadnezzar, perhaps curious, permitted a dissenting group to consume a diet of just legumes and water. After 10 days, the data was in: The legume-eaters were clearly better nourished than the meat-eaters.
Chronicled in the Bible’s Book of Daniel, Nebuchadnezzar’s experiment marks perhaps the first documented clinical trial in human history, though the evolutionary path to today’s gold standard — a randomized, double-blinded, placebo-controlled clinical trial — has required several centuries of constant change and improvement.
The COVID-19 pandemic is, in some ways, another inflection point. Among all of the other ways that the pandemic has altered how we live and think, it prompted new reassessments and reimaginings of how clinical trials can and should be conducted.
With unprecedented speed and scope, a pair of vaccines based on messenger RNA (mRNA) technology from Pfizer-BioNTech and Moderna sped through testing to receive emergency use approval (EUA) in a fraction of the time historically required. They have since inoculated more than 1 billion people around the world to prevent or reduce harm or death from infection by the SARS-CoV-2 virus that causes COVID-19.
“The pandemic demonstrated that clinical trials can be performed rapidly and safely when there is a crisis, when sufficient numbers of people are affected, and the Food and Drug Administration (FDA) and other governmental organizations recognize the urgency,” said Stephen Spector, MD, Distinguished Professor of Pediatrics in the UC San Diego School of Medicine and principal investigator for the UC San Diego arm of the Moderna trial.
“Not every study can be performed with this level of efficiency, but lessons have been learned that, when applied to current and future studies can, help to expedite the review and approval of important new investigational vaccines and drugs.”
At the height of the pandemic, the seeming alacrity with which the COVID-19 mRNA vaccines raced to EUA alarmed many, who feared the process was too rushed and the science too uncertain, though The Commonwealth Fund later estimated that the U.S. vaccination program prevented more than 1.1 million deaths and 10.3 million hospitalizations.
“Our estimates suggest that in 2021 alone, the vaccination program prevented a potentially catastrophic flood of patients requiring hospitalization. It is difficult to imagine how hospitals would have coped had they been faced with 10 million people sick enough to require admission,” wrote Commonwealth Fund authors.
“In addition to reducing hospitalization and death, vaccination prevented many millions of COVID infections, reducing the prospect that millions of Americans could develop long-COVID and its debilitating symptoms.”
The indisputable success story of the mRNA COVID-19 vaccines — and the particulars of how they were put into effect so quickly — has added fuel to ongoing debates about the nature and processes of clinical trials, and most notably how they can, should and need to change.
UC San Diego Health researchers and physicians oversee and manage hundreds of new and on-going clinical trials each year. There are more than 1,000 currently listed on its website at clinicaltrials.ucsd.edu. No institution in the region conducts more trials or across a broader spectrum of diseases, treatments and conditions. No institution is working harder to make them more effective.
“The traditional double-blind, placebo-controlled trial is still the gold standard, but alternatives are emerging.”
“The traditional double-blind, placebo-controlled trial is still the gold standard, but alternatives are emerging,” said Gary S. Firestein, MD, director of the Altman Clinical and Translational Research Institute at UC San Diego, which provides scientific, clinical, administrative and logistical support to clinical trials at UC San Diego.
(A double-blind study is one in which neither the participants nor the researchers know who is receiving treatment or a placebo. The goal is to minimize expectation bias, which leads patients or doctors to observe improvement even though a “dummy” medicine is given.)
Among the emerging alternatives and changes:
- Adaptive trials that can be altered in process as treatments and conditions change
- The use of real-world data derived from electronic health records (EHR)
- “In silico” trials based on computer simulations in combination with EHR information
- Open label trials in which both health providers and patients are aware of the drug - The use of surrogate biomarkers, such as the presence or absence of a specific - Trials under the Food and Drug Administration’s (FDA) “animal rule” when human trials are not possible

Gary S. Firestein, MD, director of the Altman Clinical and Translational Research Institute at UC San Diego
Accelerating drug development
Before a new drug or treatment can reach market, it must pass through several stages of testing and assessment, beginning with preclinical research, which alone can take years, sometimes decades. One reason the mRNA COVID-19 vaccines could be put into clinical trials was the fact that the approach had already been the subject of years of research and testing for other diseases.
Generally speaking, it takes seven to 10 years for a clinical trial to successfully move through all four phases (see sidebar), though longer is not uncommon. Many attempts have been made to shorten the process.
In 2016, for example, Congress passed the 21st Century Cures Act, partly aimed at streamlining drug and device approval. Some public officials and others, then and now, argued for simply truncating trials, suggesting that promising new drugs could be released to the public after satisfying only basic safety requirements.
In 2017, Jim O’Neill, a former official at the U.S. Department of Health & Human Services declared, “Let’s prove efficacy after they’ve been legalized.”
Most experts don’t go that far.
“Shortening clinical trials has the potential to move interventions through the study phase too quickly. Interventions may have outcomes that cannot be detected in a trial that lasts weeks rather than years,” said Anthony Magit, MD, professor of surgery and medical director of the Office of IRB Administration at UC San Diego.
“With innovative study design, including better characterization of subjects, fewer subjects may need to be enrolled, but longer studies may be necessary to assess a broader range of outcomes. Long-term follow-up after approval for clinical care may be an important component to evaluate sustained benefit and to monitor for negative outcomes.”
Clinical trials for the breast cancer drug tamoxifen, for example, began in the 1970s; late-stage trials are ongoing.
Some experts have advocated for modifying the third phase of clinical trials, which they deem unnecessarily time-consuming, expensive and redundant. They favor “adaptive licensing,” especially for promising therapeutics that address critical or urgent needs, such as new vaccines, antibiotics, cancer medications or therapies for Alzheimer’s disease.
These drugs would be rolled out sooner and faster, beginning with limited distribution to the sickest patients after conclusion of Phase I. Early recipients would be monitored and, if the drug appeared safe and effective, availability would be expanded to more categories of patients, a process dubbed “progressive reduction of uncertainty.”
One problem is that safety signals might not be seen in limited trials, said Firestein, and effectiveness is not assured. This can create issues with third-party payers who bear the cost of expensive drugs that might not work.
Sandip Patel, MD, associate professor at UC San Diego School of Medicine and director of the clinical trials office at Moores Cancer Center at UC San Diego Health is among those advocating for modified trials.
“In cancer clinical trials, there is a movement toward emphasizing novel therapeutic intent over a historical emphasis on statistical balance, which has improved access to trials,” Patel said.
“With the current era focusing on approvals of novel therapeutics from Phase I in record time to help patients, such as the COVID-19 efforts, and continued vigilance throughout a drug’s life cycle in both trials and real-world use, I think the old model of Phase I to Phase II to Phase III as discrete elements is a relic of the past.”
At the HUMANOID Center of Research Excellence, the translational arm of the Institute for Network Medicine at UC San Diego School of Medicine, founding co-directors Soumita Das, PhD, and Pradipta Ghosh, MD, with staff director Courtney Tindle, talk about another way to tweak clinical trials. It starts with “Phase 0.”
The current multi-phase is arduous, but not necessarily productive. Most drugs fail somewhere along the pipeline, observed Das, many in the last phase because of a primary lack of efficacy. In other words, they never had a chance.
The HUMANOID approach leverages human organoids to model “diseases-in-a-dish.” These multi-dimensional, multi-cell-type mini-organs can be a more realistic replication of complex human tissues than single cell cultures or most animal models. (See page 68 for more on organoids.) If an experimental compound doesn’t work in an organoid (Phase 0), it can be jettisoned with less waste of time and resources, said Das.
“HUMANOID’s organoid-based models provide a head-start on efficacy, giving researchers an incredible tool to separate useful therapies from dead ends. This is how we accelerate the drug discovery process and bring new hope to patients.”
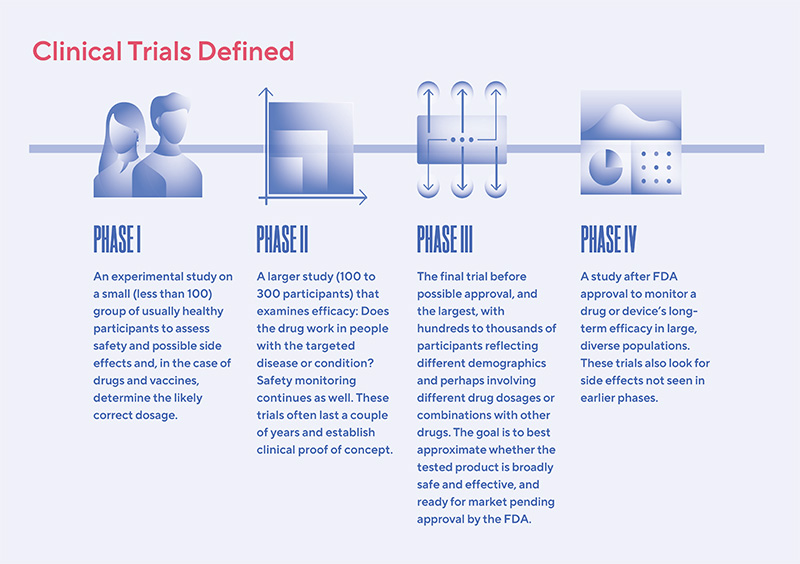
Clinical trials are designed to evaluate whether a new drug or medical device is safe and effective.
Fixing problems (at least some of them)
Problem #1: Diversity
The history of medical research is fraught with stories of women and minorities either underrepresented or abused, i.e. the notorious Tuskegee study in which researchers studied the effects of untreated syphilis in Black men over the course of 40 years.
By some estimates, as much as 90 percent of clinical trials participants have historically been white and of European descent, though disease clearly affects different people differently. A 2022 survey of more than 20,000 U.S.-based trials from 2000 to 2020 with published results found that less than half reported any race/ethnicity data. The majority of enrollees (mean 79.7 percent) were White, with all other race/ethnic groups underrepresented compared to census estimates.
“There is a push now to involve multiple sites and be inconclusive with respect to racial, ethnic and socioeconomic diversity to represent the diversity in society,” said Magit. “A critical component of representing as much of society as possible is providing gender equity in clinical trial recruitment, as clinical trials have historically overrepresented males. One overall goal is to conduct clinical trials where the results are directly translatable to the affected population, rather than having to make adjustments due to narrow inclusion criteria or only including a subset of the affected population.”
Diversity was an essential driver of the COVID-19 vaccine trials. The disease impacted people everywhere, but in the United States at least, it struck minority groups especially hard, with higher rates of hospitalization and death.
Spector noted that, with great effort, of the 336 study participants enrolled in the Moderna vaccine trial at UC San Diego, more than 50 percent represented minorities and persons of color.
Susan Little, MD, professor of medicine at UC San Diego School of Medicine and principal investigator for the UC San Diego study arms for two international COVID-19 vaccine trials (Astra-Zeneca and Johnson and Johnson) said the emphasis in both trials was increased participation in underserved communities, particularly those of color. These populations are traditionally underrepresented in clinical trials and remain overrepresented in rates of SARS-CoV-2 disease, hospitalization and death.
One visible way to do that, she said, was to create mobile clinics, including specially equipped vehicles, that brought vaccination opportunities to communities with residents unlikely or unable to visit the usual university-based testing sites. The clinics were often supported by local health care providers, adding familiarity and trust to the process.
“The SARS-CoV-2 pandemic has disproportionately impacted communities of color across the United States,” said Little. “These vehicles help our research team bring vaccine trial opportunities to high-burden communities that might otherwise be underserved.”
Problem #2: Getting data
In 2014, Thomas Insel, MD, director of the National Institute of Mental Health (NIMH) from 2002 to 2015, noted that one of the reasons mental illness was so poorly understood is the lack of knowledge about the underlying brain mechanisms involved, including how they worked or malfunctioned.

UC San Diego used mobile clinics to bring vaccine trial opportunities to high-burden communities that might otherwise be underserved and underrepresented in clinical trials.
Insel introduced a new NIMH policy that required clinical trials of psychiatric therapies to include basic research. That way, even if the trial failed, researchers might at least learn something about how the brain works.
Since his arrival at UC San Diego in 2016 to head the Alzheimer’s Disease Cooperative Study (ADCS), Howard Feldman, MD, professor of neuroscience, has redirected efforts away from high-profile, large, late stage clinical trials in Alzheimer’s disease (which so far have had an extremely high failure rate and which have not resulted in an approved, market-ready, payer-supported treatment) to smaller, more nimble studies directed at achieving clinical proof of concept as a strategy to buy down risk of later stage costly failures.
“At UC San Diego, we are ideally suited for collaborative, academic-led clinical trials taking on studies that are unlikely to be done by industry.”
“We strive to keep focused on the highest quality research and achievable research goals that answer important questions with approaches that will make a difference in the lives of our patients,” said Feldman.
“At UC San Diego, we are ideally suited for collaborative academic led clinical trials, taking on studies that are unlikely to be done by industry, or where ADCS might be an ideal partner. By contributing earlier stage trials each step successfully completed helps move us closer to our goal of benefiting persons at risk or with Alzheimer’s disease.”
Problem #3: Sharing data
In 2007, the United States became the first nation to launch a database of clinical trial results involving drugs, devices and biologics, such as vaccines. The law described significant penalties for failure to comply, such as withholding of federal funding. The goal was to encourage sharing of information, especially if a trial failed, so that future research would be better informed.
It hasn’t quite worked out as envisioned, even with a 2017 update in which the National Institutes of Health (NIH) and FDA enacted a “final rule” clarifying expectations and penalties. The results of many clinical trials remain unknown, especially for those that fail or fall short.
In 2020, for example, Science examined more than 4,700 trials whose results should have been posted to the ClinicalTrials.gov website. Reporting rates improved after the 2017 rule, but the journal still reported that hundreds of trials had not released data.
That’s perhaps not surprising given the overall likelihood of a clinical trial successfully moving from Phase I to market approval is less than 10 percent, with almost 70 percent of trials not making the jump from Phase II to Phase III.
“No one gets excited about publishing a negative result, and it is difficult to publish them in top journals,” said Firestein, who noted, too that trials that struggle or fail to accrue sufficient numbers of participants often lead to underpowered and suspect results.
Recruitment is an almost universal challenge. Eighty percent of trials are delayed or closed because of difficulties finding participants. Nine out of 10 trials double their original timeline in order to meet enrollment goals. One in 10 trial sites fails to enroll a single participant.
Problem #4: Getting drugs and vaccines to children
The COVID-19 vaccines demonstrated a common medical challenge: Approving life-saving vaccines and drugs for children. As of 2018, according to a study published in Health Affairs, 64 percent of new drugs were not approved for children, despite laws and policies specifically intended to promote their development.
In fact, more than 30 percent of all prescriptions written for children involve off-label medications — the practice of using a drug to treat a disease or medical condition that it is not formally approved to treat.
“There is this fear that children are too fragile to be involved in clinical trials prior to FDA approval first in adults,” said Spector. “However, for serious diseases, drugs can be studied in parallel in children and adults once Phase I data have demonstrated the safety and pharmacokinetic properties of the drug under investigation.
“The marked increase in infections among unimmunized children during the Delta and Omicron waves of COVID-19 demonstrate that when children are not included in the original efficacy trials, they are unnecessarily put at continued risk for disease.”
The FDA approved EUAs for mRNA COVID-19 vaccines for children ages 5 to 11 in October 2021, almost one year after the first EUA for adults. An EUA for children between 5 and six months was granted in June 2022.

Howard Feldman, MD, neurologist and professor of neurosciences at UC San Diego School of Medicine
The message of mRNA
Messenger RNA was discovered in the early 1960s, a single strand of ribonucleic acid that is complementary to one of the two strands of DNA in a gene and which provides the blueprint for making proteins. mRNA does not, however, change DNA (an unfounded fear voiced by some early COVID-19 vaccine opponents) and rapidly decays.
Within a decade, scientists were experimenting with using droplets of fat called liposomes to transport mRNA into cells, where the latter might induce very specific protein expression for targeted therapies.
It would take another decade or so before mRNA would be tested as a vaccine (for influenza in mice). By the 2000s, mRNA vaccines were being studied for infectious diseases ranging from rabies to Zika, though technical and logistical challenges proved daunting. Both Pfizer’s and Moderna’s vaccines, for example, must be stored at sub-zero temperatures to protect the fragile lipid droplets.
The global urgency of COVID-19 dramatically altered the situation. No effort could be spared. Every potential vaccine needed to be explored, no matter the challenges or costs.
Messenger RNA vaccines for COVID-19 have proven their value. They are faster and cheaper to develop than traditional approaches, and their success has spurred renewed and expanded interest in using the technology for malaria, tuberculosis, hepatitis B and cystic fibrosis, plus mRNA treatments for several types of cancer.
“The COVID-19 vaccines have demonstrated that mRNA technology is safe and has the potential to be used for many other vaccines and novel therapies,” said Spector. “But they also identified a potential limitation of the technology, if it turns out that mRNA vaccines only induce short-term immunity (rather than it being a consequence of always-evolving viruses). So although mRNA technology has overcome major safety and regulatory hurdles, the long-term benefit of this technology still requires evaluation and further refinement.”
This first batch of COVID-19 vaccines was received by UC San Diego Health in December 2020.
N=1
Clinical trials are often not definitive. A drug that successfully reaches market may prove problematic later because of unanticipated side effects. Cholesterol-reducing statins, for example, are among the most-researched and most-prescribed drugs in the world. An estimated 60 percent of Americans over the age of 65 take a statin to help prevent a heart attack or stroke. More than 200 million people take statins worldwide.
But Phase IV trials and other research has found statins can cause blood sugar abnormalities, liver damage, debilitating muscle pain or neurological issues, including memory problems and cognitive decline in some people. These adverse effects, which were considered rare or undetected in clinical trials, took years and many users to fully reveal themselves.
Conversely, noted Firestein, international clinical trials of the Alzheimer’s drug aducanumab (Aduhelm) produced disappointing results and were halted in 2019. Subsequent retrospective assessments by the drugmaker suggested Aduhelm benefitted some patients. The FDA, surprisingly and controversially, approved it based on urgent need despite overwhelming opposition by its own expert advisors. The Centers for Medicare and Medicaid Services later ruled that the drug was only covered for patients in approved clinical trials.
Randomized clinical trials work best at predicting treatments benefitting the average patient, but most diseases and conditions play out in unique ways in each individual affected. There is no such thing as an “average patient.”
In statistics, the symbol “n” represents the total number of individuals or observations in a sample. The “n” in the Phase III trial for Pfizer-BioNTech’s COVID-19 vaccine, for example, equaled 43,661 participants.
The concept of N=1, or testing a drug on a single individual, dates back at least to 1986 and the description of a one-patient experiment in The New England Journal of Medicine. That singular trial was successful: Researchers identified a specific asthma treatment for one patient. The trial spurred greater interest, but N=1 trials remain more anomaly than trend.
“N=1 is intellectually appealing, especially for rare diseases,” said Firestein, “but they are hard to do, expensive and the results often cannot be generalized for the general good. I usually see them as hypothesis-generating so that a properly controlled study can be performed.”
Clinical trials in the future
Ask a researcher what should or could be changed to improve the efficacy of clinical trials, and their answers are likely to be N=Many. Much has already been accomplished, such as tapping into electronic health records to identify potential participants rather than leaning on doctors to find eligible patients or leveraging communications tools like social media and advertising.
Clinical trials require approval by institutional review boards (IRB) to ensure the rights and welfare of human research subjects are protected. In recent years, there has been a shift to central IRBs, as opposed to boards at each institution, especially for multi-site studies. Central IRBs have streamlined the process by creating a single, umbrella approval for many studies.
But Firestein focuses on one particular improvement: Is a proposed trial feasible? Are there patients that meet the entry criteria? If not, can it actually be done?
“We have wanted to do this for several years, but investigators push back, saying that it interferes with academic freedom. But a study that is not feasible because the patients don’t exist doesn’t help anyone.”

About Discoveries Magazine
This article was originally featured in the 2023 issue of Discoveries, the annual magazine from UC San Diego Health Sciences. Discoveries highlights the latest and greatest in the university's health science research, education and clinical practice. Other stories in this issue explore topics such as mindfulness meditation, wearable medical devices and a profile of new Vice Chancellor for Health Sciences, John M. Carethers.
Read more news about: Health Sciences
Share This:
You May Also Like




Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.