UC San Diego Team Aims to Broaden Researcher Access to Protein Simulation
Researchers Enable Unprecedented Sampling Using Desktop Computers with Off-the-Shelf Upgrade
By:
Published Date
Article Content
Using just an upgraded desktop computer equipped with a relatively inexpensive graphics processing card, a team of computer scientists and biochemists at the University of California, San Diego, has developed advanced GPU accelerated software and demonstrated for the first time that this approach can sample biological events that occur on the millisecond timescale.
These results have the potential to bring millisecond scale sampling, now available only on a multi-million dollar supercomputer, to all researchers, and could significantly impact the study of protein dynamics with key implications for improved drug and biocatalyst development.
With some innovative coding, a GPU (graphics processing unit) that retails for about $500, and the widely used software package of molecular simulations called Amber (Assisted Model Building with Energy Refinement), the researchers were able to run a simulation showing the same five long-lived structural states of a specific protein as observed in a simulation conducted by D.E. Shaw Research’s Anton, a purpose-built molecular dynamics (MD) supercomputer. The Anton simulation was conducted over a period of slightly more than one millisecond – or 100 times longer than the previous record.
“This work shows that using conventional, off-the-shelf GPU hardware combined with an enhanced sampling algorithm, events taking place on the millisecond time scale can be effectively sampled with dynamics simulations orders of magnitude shorter (2000X) than those timescales,” the researchers wrote in their paper, ‘Routine Access to Millisecond Timescale Events with Accelerated Molecular Dynamics.’ The paper was published July 27 in the Journal of Chemical Theory and Computation and can be viewed here.
The enhanced sampling algorithm refers to the use of accelerated molecular dynamics, or aMD, a method that improves the conformational space sampling of proteins when compared with conventional molecular dynamics simulations, or cMD.
Specifically, the UC San Diego researchers analyzed the bovine pancreatic trypsin inhibitor (BPTI), a small protein with 58 residues. BPTI was the first protein to be simulated, in 1977, with J. Andrew McCammon, the Joseph E. Mayer Chair of Theoretical Chemistry at the UC San Diego, as lead author on that milestone research.
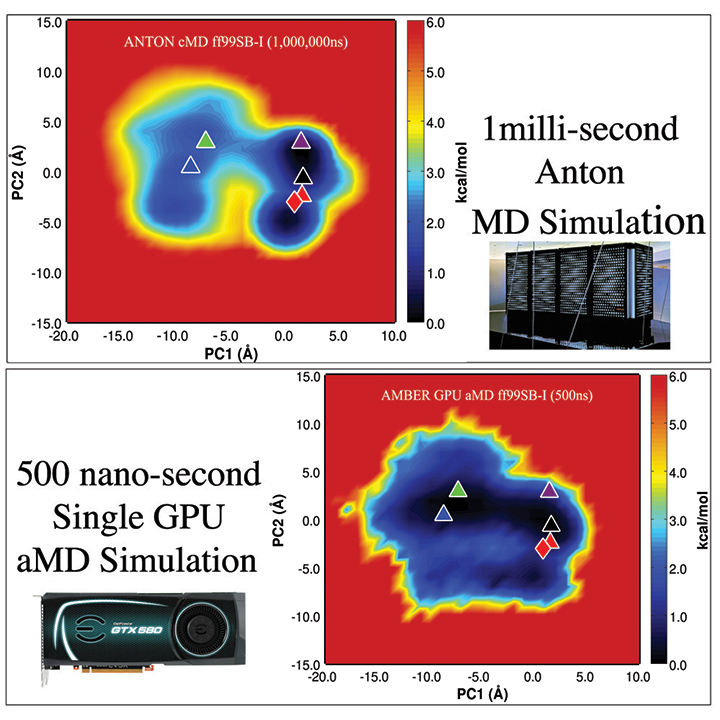
Images showing the conformational space explored by the protein in the 1ms conventional MD Anton simulation, and the 500ns aMD simulation. The red diamond marks the crystal structure where both simulations were started from; the triangles represent important structures found in the 1ms simulation. The lower image shows that with a single graphics card running 500ns of accelerated MD, the same structures can be sampled and the same relative conformational space can be explored. Principal Component Analysis (PCA) captures the slowest motions of the protein, which correspond to conformational changes on long timescales. Moving L to R along the x-axis captures the primary slow rocking motion of the protein; moving vertically on the y-axis captures the second slowest motion, or wagging movement. Courtesy of the Walker MD Lab, San Diego Supercomputer Center.
“The breakthrough described in the new paper was achieved by combining advances in theory and in computer technology, but other types of resources such as SDSC’s new Gordon supercomputer are also increasingly needed for large, data-intensive simulations,” said McCammon, part of the research team on the latest findings. McCammon is also a chemistry and biochemistry professor in UC San Diego’s Division of Physical Sciences, a Distinguished Professor of Pharmacology at UC San Diego, Investigator of the Howard Hughes Medical Institute, and a Fellow with the university’s San Diego Supercomputer Center (SDSC).
While the team’s aMD simulation was only 500 nanoseconds long, or .0005 of a millisecond, the group was able to sample all of the structural states seen in the longer timescale simulation run on Anton.
“In just 500 nanoseconds, we saw the same things as in the Anton simulations, which we used as an excellent benchmark,” said Romelia Salomon-Ferrer, an SDSC postdoctoral research fellow and member of the team who ported aMD to Amber. “We were able to cover that same space faster. One could compare that to having a choice between taking a train or a plane to San Francisco. The distance is the still the same; however the plane would get there faster. But this would also be a very particular plane in the sense that it is also relatively inexpensive.”
In addition to potentially broadening access among researchers by enabling desktop simulations, the UC San Diego research also marks the longest aMD simulation of a biomolecule to-date, as well as the first “apples-to-apples” comparison of an aMD simulation versus a very long cMD simulation.
“The key to this work has been to sit down and rethink the problem from the beginning,” said Ross Walker, an assistant research professor with the SDSC, principal investigator (PI) and corresponding author of this research. “We had already massively accelerated conventional MD on GPUs but even this was not going to be sufficient to allow us to routinely sample conformational events taking place on the millisecond timescale. By combining our experience with conventional MD on GPUs with the enhanced sampling provided by accelerated MD methods, we were able to exploit both providing, for the first time, the ability to routinely simulate events that take place on the millisecond timescale.”
“Furthermore, GPUs offer the potential for supercomputing performance on the average desktop computer, giving researchers the ability to test multiple hypotheses in real time,” said Walker, who also is an adjunct assistant professor in UC San Diego's Department of Chemistry and Biochemistry, and an NVIDIA CUDA Fellow. “The NSF-funded work we are doing in the Walker Molecular Dynamics Lab at SDSC to develop GPU accelerated software promises to transform how scientists approach applying molecular dynamics techniques that may ultimately lead to the design of new drugs and biological catalysts.”
“Running the entire MD simulation on the GPU as opposed to other approaches has really allowed us to run them much more efficiently, both in terms of conventional MD and now accelerated MD,” said Levi C.T. Pierce, lead author of the paper and a postdoctoral research fellow with SDSC and the university’s Department of Chemistry and Biochemistry. “The conventional MD in Amber was completely rewritten to run on the GPU, while the enhanced sampling method, aMD, has been coded into the GPU, allowing us to access these long time scale dynamics.”
The researchers, however, cautioned that while aMD is very useful for the exploration of conformational space – the different structures explored as the protein fluctuates – it does not reproduce the exact timescale of these fluctuations.
“Accelerated molecular dynamics may not solve the entire problem, but it is a really good initial tool,” said Salomon-Ferrer. “By using aMD along with Amber, we can lower the financial and logistical barriers to research, and one particularly important characteristic of aMD is that researchers don’t really need to know anything about the complexities of a specific protein beforehand.”
Also participating in the research was Cesar Augusto F. de Oliveira, with the UC San Diego Department of Chemistry and Biochemistry and the Howard Hughes Medical Institute.
Researchers used GPU desktops in Walker’s lab as well as a GPU compute cluster at SDSC and the resources of the Keeneland computing facility at the Georgia Institute of Technology. The work was funded in part by the National Science Foundation (NSF) through its Scientific Software Innovations Institutes program - NSF SI2-SSE (NSF1047875 & NSF1148276) grants, the NSF’s Extreme Science and Engineering Discovery Environment (XSEDE) program, and by a University of California grant (UC Lab 09-LR-06-117792).
Computer time was provided by SDSC through NSF award TGMCB090110. The research was also supported by Walker’s CUDA fellowship from NVIDIA. The J. Andrew McCammon Group is supported by the NSF, National Institutes of Health (NIH), Howard Hughes Medical Institute (HHMI), National Biomedical Computation Resource (NBCR), and Center for Theoretical Biological Physics (CTBP).
Share This:




Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.