‘Leaky’ Activity of Mutated Enzyme Underlies Neurodegenerative Disease
UC San Diego study finds enhanced PKCγ activity drives spinocerebellar ataxia type 14; suggests inhibiting the enzyme may have therapeutic potential
Story by:
Published Date
Article Content
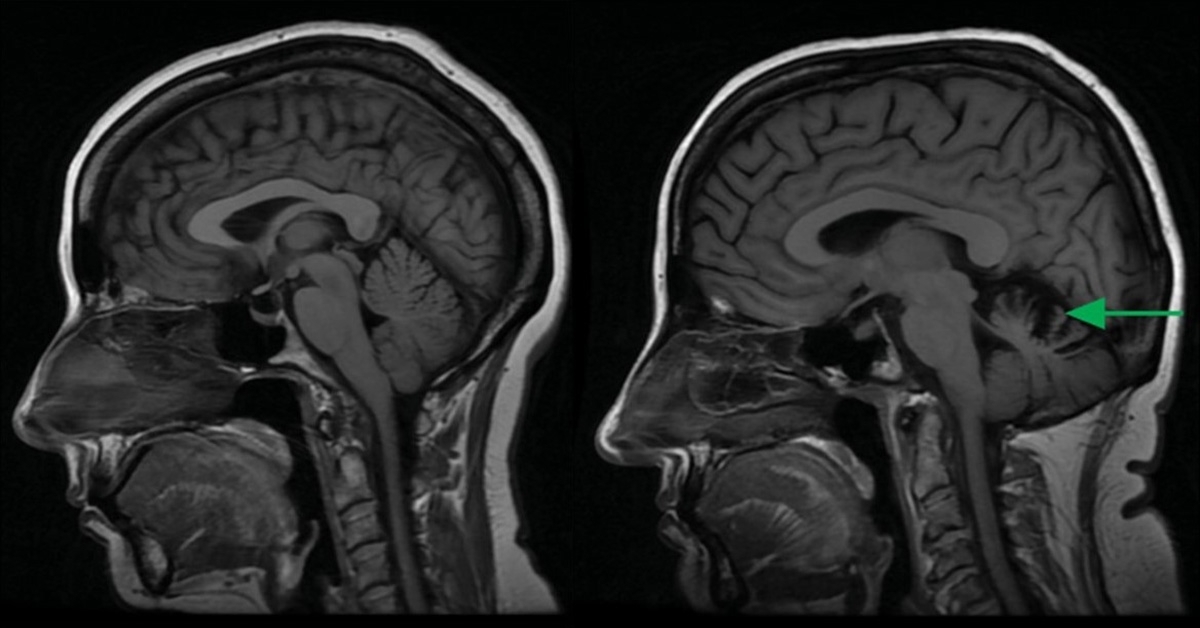
Spinocerebellar ataxias are a group of neurodegenerative diseases characterized by the degeneration of Purkinje cells, a major class of neurons in the cerebellum. The resulting cerebellar dysfunction leads patients to experience a loss of motor coordination and control.
One subtype of the disease, spinocerebellar ataxia type 14 (SCA14), was found to be caused by mutations in protein kinase C-gamma (PKCγ), an enzyme that regulates other proteins in Purkinje cells. But exactly how these mutations alter the enzyme’s function to ultimately drive neurodegeneration remained unknown.
In a new study, published September 27, 2022 in Science Signaling, researchers at University of California San Diego School of Medicine found that SCA14-associated mutations disrupt the autoinhibition and degradation of PKCγ, leading to elevated levels of enzyme activity. This sustained “leaky” activity alters the Purkinje cell phosphoproteome to drive cerebellar pathology.
“Our findings reveal important mechanisms underlying spinocerebellar ataxia and position PKCγ as a promising therapeutic target for this neurodegenerative disease,” said senior author Alexandra C. Newton, PhD, Distinguished Professor of Pharmacology at UC San Diego School of Medicine.
To understand how the SCA14-associated mutations affect the enzyme’s function, researchers first measured activity levels of different PKCγ variants in cultured cells. Compared to more common PKCγ variants, those with SCA14 mutations in the protein’s C1A and C1B domains showed significantly enhanced enzymatic activity, which further experiments confirmed was due to conformational changes that impair the enzyme’s autoinhibition and degradation.
Autoinhibition is an on-site regulatory mechanism in which certain domains within a molecule’s structure act to repress its own function.
Researchers then found the enhanced PKCγ activity led to a cascade of downstream changes to the phosphorylation state of the cellular environment, particularly dysregulating signaling pathways involved in axon development and cytoskeletal structure.
The extent of disrupted PKCγ autoinhibition correlated with disease severity, and mutations that induced a particularly high level of PKCγ activity were also associated with an earlier age of disease onset.
PKCγ is itself regulated by intracellular calcium, and many other types of spinocerebellar ataxia are driven by mutations that affect calcium homeostasis. Thus, the authors suggest that targeting PKCγ may correct this broader signaling pathway and prove effective in treating multiple forms of the disease.
“This raises exciting possibilities for therapeutically targeting PKCγ not only in SCA14 but also in many other subtypes of spinocerebellar ataxia,” Newton said.
Co-authors of the study include: Caila A. Pilo, Timothy R. Baffi, Alexandr P. Kornev, Maya T. Kunkel, Mario Malfavon, Leigh-Ana Rossitto, David J. Gonzalez and Susan S. Taylor at UC San Diego; Dong-Hui Chen, Daniel X. Chen and Wendy H. Raskind at University of Washington; Liang-Chin Huang and Natarajan Kannan at University of Georgia; and Cheryl Longman and George Gorrie at Queen Elizabeth University Hospital.
The study was funded, in part, by the National Institutes of Health (grants R35GM122523, R01NS069719 and R35GM139656), the Zionic Ataxia Fund and the National Ataxia Foundation.
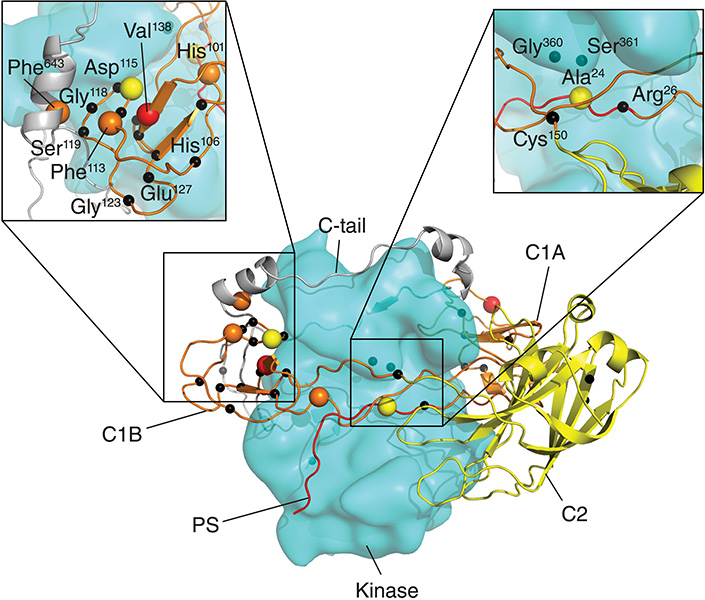
A model of PKCγ with spheres indicating SCA14-associated mutations.
Share This:
You May Also Like




Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.