DNA Treatment Could Delay Paralysis That Strikes Nearly All Patients with ALS
In both mouse and human motor neuron studies, a DNA designer drug restored levels of a protein necessary to keep motor neurons functioning, returning activity impaired in amyotrophic lateral sclerosis; findings could lead to clinical trials.
Story by:
Published Date
Article Content
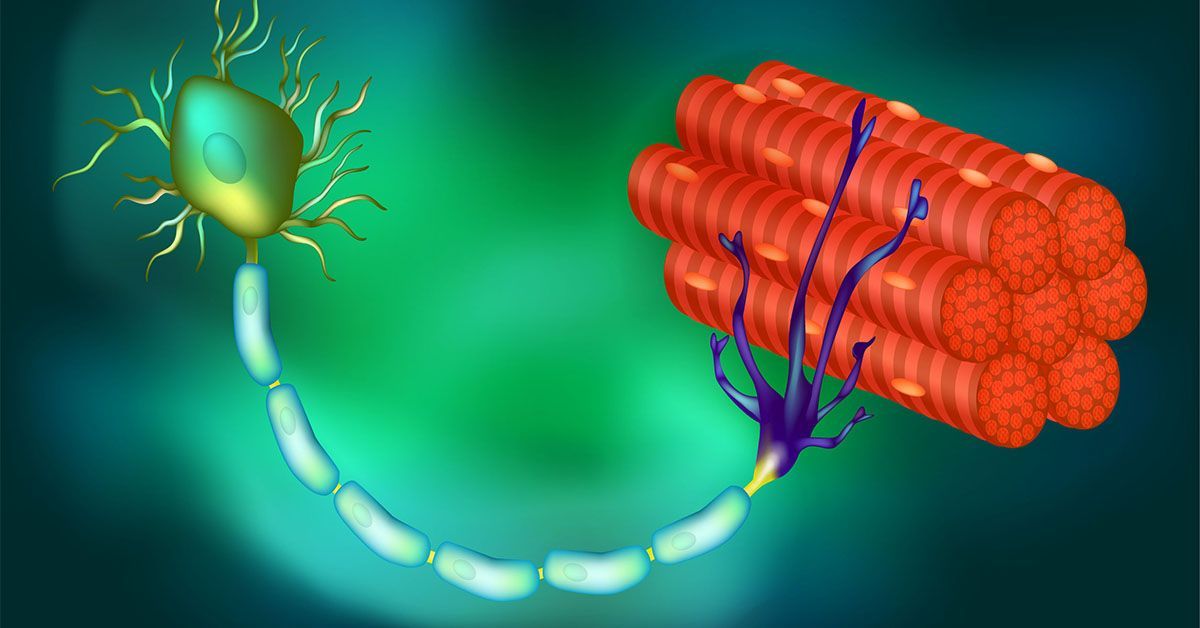
In virtually all persons with amyotrophic lateral sclerosis (ALS) and in up to half of all cases of Alzheimer’s disease (AD) and frontotemporal dementia, a protein called TDP-43 is lost from its normal location in the nucleus of the cell. In turn, this triggers the loss of stathmin-2, a protein crucial to regeneration of neurons and the maintenance of their connections to muscle fibers, essential to contraction and movement.
Writing in the March 16, 2023 issue of Science, a team of scientists, led by senior study author Don Cleveland, PhD, Distinguished Professor of Medicine, Neurosciences and Cellular and Molecular Medicine at University of California San Diego School of Medicine, with colleagues and elsewhere, demonstrate that stathmin-2 loss can be rescued using designer DNA drugs that restore normal processing of protein-encoding RNA.
“With mouse models we engineered to misprocess their stathmin-2 encoding RNAs, like in these human diseases, we show that administration of one of these designer DNA drugs into the fluid that surrounds the brain and spinal cord restores normal stathmin-2 levels throughout the nervous system,” Cleveland said.
Cleveland is broadly credited with developing the concept of designer DNA drugs, which act to either turn on or turn off genes associated with many degenerative diseases of the aging human nervous system, including ALS, AD, Huntington’s disease and cancer.
Several designer DNA drugs are currently in clinical trials for multiple diseases. One such drug has been approved to treat a childhood neurodegenerative disease called spinal muscular atrophy.
The new study builds upon ongoing research by Cleveland and others regarding the role and loss of TDP-43, a protein associated with ALS, AD and other neurodegenerative disorders. In ALS, TDP-43 loss impacts the motor neurons that innervate and trigger contraction of skeletal muscles, causing them to degenerate, eventually resulting in paralysis.
“In almost all of instances of ALS, there is aggregation of TDP-43, a protein that functions in maturation of the RNA intermediates that encode many proteins. Reduced TDP-43 activity causes misassembly of the RNA-encoding stathmin-2, a protein required for maintenance of the connection of motor neurons to muscle,” said Cleveland.
“Without stathmin-2, motor neurons disconnect from muscle, driving paralysis that is characteristic of ALS. What we have now found is that we can mimic TDP-43 function with a designer DNA drug, thereby restoring correct stathmin-2 RNA and protein level in the mammalian nervous system.”
Specifically, the researchers edited genes in mice to contain human STMN2 gene sequences and then injected antisense oligonucleotides — small bits of DNA or RNA that can bind to specific RNA molecules, blocking their ability to make a protein or changing how their final RNAs are assembled — into cerebral spinal fluid. The injections corrected STMN2 pre-mRNA misprocessing and restored stathmin-2 protein expression fully independent of TDP-43 function.
“Our findings lay the foundation for a clinical trial to delay paralysis in ALS by maintaining stathmin-2 protein levels in patients using our designer DNA drug,” Cleveland said.
Co-authors include: Michael W. Baughn, Jone López-Erauskin, Melinda S. Beccari, Roy Maimon, Sonia Vazquez-Sanchez, Jonathan W. Artates and Eitan Acks, all at Ludwig Institute for Cancer Research-UC San Diego and UC San Diego; Ze’ev Melamed, Ludwig Institute for Cancer Research-UC San Diego, UC San Diego, and The Hebrew University of Jerusalem; Karen Ling, Paayman Jafar-nejad, Frank Rigo and C. Frank Bennett, all at Ionis Pharmaceuticals; Aamir Zuberi, Maximilliano Presa, Elena Gonzalo-Gil and Cathleen Lutz, all at The Jackson Laboratory; Som Chaturvedi, Mariana Bravo-Hernández, Vanessa Taupin and Stephen Moore, all at UC San Diego; L. Sandra Ndayambaje and Ana R. Agra de Almeida Quadros, Harvard Medical School; Clotilde Lagier-Tourenne, Harvard University and Broad Institute of Harvard University and Massachusetts Institute of Technology.
Funding for this research came, in part, from the National Institutes of Health (grants R01NS112503, RF1NS124203), ALS Finding a Cure, Massachusetts Center for Alzheimer Therapeutic Science, The Sean M. Healey & AMG Center for ALS at Mass General, U42 Mutant Mouse Resource Research Center (OD010921), Ruth Kirschstein Institutional National Research Service Award (T32 GM008666 and T32 AG 66596-2), The Packard Center for ALS Research, The ALS Association, MDA development grants, The BrightFocus Foundation and Cancer Center Support Grant (CA034196).
Disclosure: Cleveland and Melamed are authors on U.S. patents describing methods and compounds for restoration of STMN2 in TDP-43 proteinopathies and a consultant for Ionis Pharmaceuticals.

Don Cleveland, PhD, Distinguished Professor of Medicine, Neurosciences and Cellular and Molecular Medicine at UC San Diego School of Medicine, is among the most highly cited researchers in the world for his work investigating neurodegenerative diseases. Erik Jepsen/UC San Diego
Share This:
You May Also Like



Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.