Big Data for Chemistry
New method helps identify antibiotics in mass spectrometry datasets
Published Date
Article Content
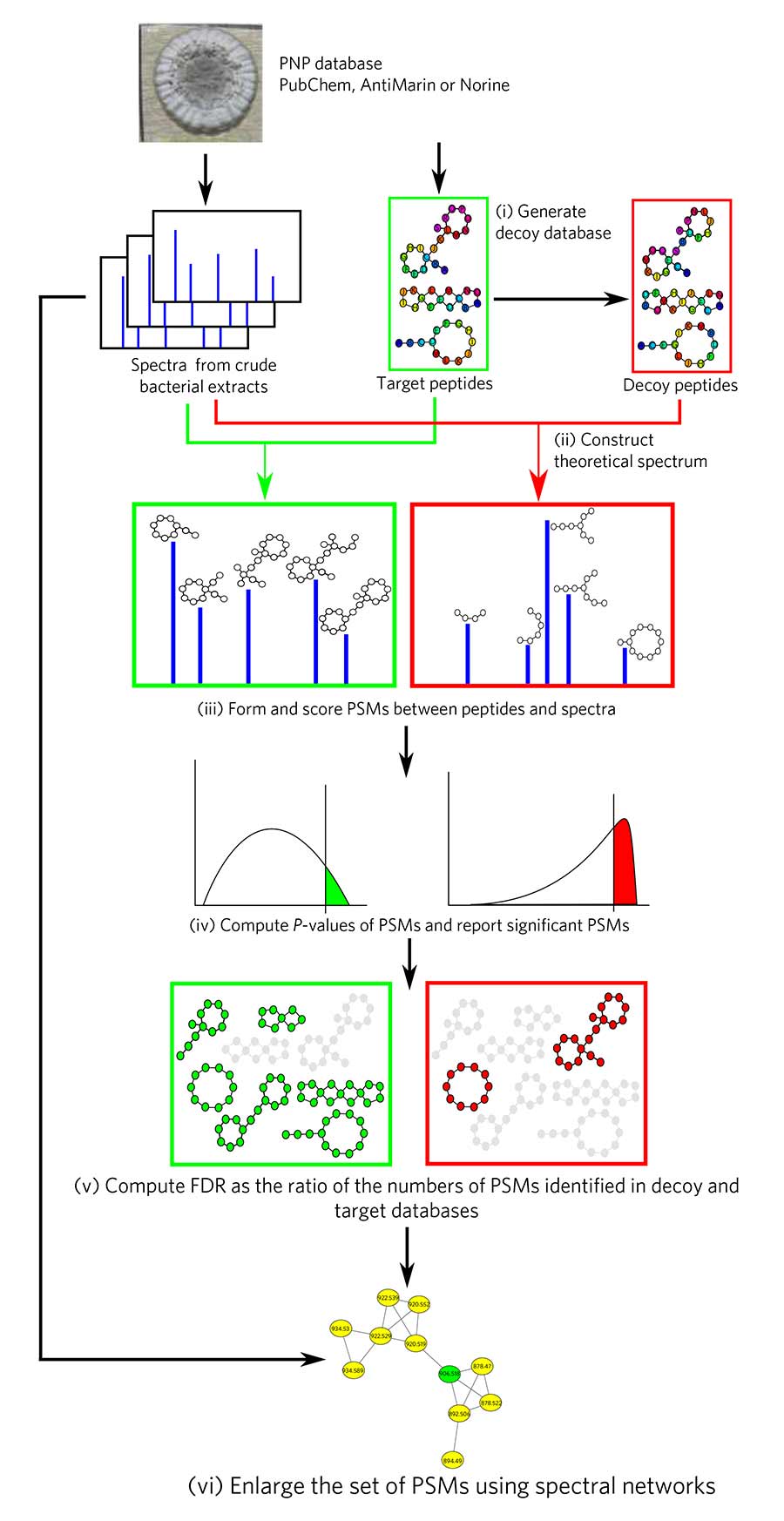
The DEREPLICATOR pipeline.
An international team of computer scientists has for the first time developed a method to find antibiotics hidden in huge but still unexplored mass spectrometry datasets. They detailed their new method, called DEREPLICATOR, in the Oct. 31 issue of Nature Chemical Biology.
Each year more than 2 million people develop antibiotic resistance in the United States, and researchers hope their work will help identify new antibiotics to effectively treat diseases.
“This is the first time that we are using Big Data to look into microbial chemistry and characterize antibiotics and other drug candidates,” said Hosein Mohimani, a computer scientist at the University of California San Diego and the paper’s first author. “Although proteomics researchers have been routinely using huge spectral datasets to find important peptides, all traditional proteomics tools fail when it comes to new drug discovery. “
The algorithms the researchers developed scour mass-spectrometry data to discover so-called peptidic natural products (PNPs)—widely used bioactive compounds that include many antibiotics.
Mass spectrometry allows researchers to identify the chemical structure of a substance by separating its ions according to their mass and charge. By running mass spectrometry data against a database of chemical structures of known antibiotics, the researchers were able to detect known compounds in substances that had never been analyzed before.
This is the first time that this kind of Big Data analysis was possible. The researchers were able to get around the well-known issue of false positives by using statistical analysis to determine the significance of each match between spectra and the antibiotics database. “We got the idea from particle physics,” Mohimani said. Researchers used a statistical approach called the Markov Chain Monte Carlo to compute the probability of rare events and to throw false positives out.
Researchers also were able to discover new variants of known antibiotics. They did that by first predicting the fragmentation pattern of a chemical structure by using chemical expertise and machine learning. They compared these predictions against experimental data and looked for patterns. This problem resembles guessing the meaning of a sentence in a foreign language by recognizing a few of the words.
A global network for mass spectrometry data
Researchers have made breakthroughs recently in antibiotics discovery, but PNPs have remained difficult to find. That’s because they’re more complex than most peptides and built from hundreds of non-standard amino acids, rather than the standard 20. As a result, standard peptide identification tools, such as SEQUEST (the workhorse of modern proteomics) do not work to identify PNPs.
The recent launch of the Global Natural Product Social (GNPS) molecular network in 2015 brought together over a hundred laboratories that have already generated an unprecedented amount of mass spectra including antibiotics. But to go from PNP discovery in an academic setting to a high-throughput technology, new algorithms for antibiotics discovery are needed. Indeed, although spectra in the GNPS molecular network represent a gold mine for future discoveries, their interpretation remains a bottleneck. The network was developed by Nuno Bandeira, a computer science professor at the Jacobs School of Engineering and study co-author Pieter Dorrestein, a professor in the UC San Diego School of Medicine and Skaggs School of Pharmacy and Pharmaceutical Sciences.
Finding complex peptides
Antibiotics researchers use dereplication strategies that identify known PNPs and discover their still unknown variants by comparing billions of spectra with a database of all known PNPs. DEREPLICATOR promises to turn into an equivalent of SEQUEST for antibiotics discovery and, similarly to SEQUEST, enable high-throughput PNP identification. Even in the first application, it identified an order of magnitude more PNPs than any previous dereplication efforts.
The study was made possible by the bioinformatics expertise in the research group of Professor Pavel Pevzner, in the Department of Computer Science and Engineering at UC San Diego who developed viable methods to sequence bacteria and metagenomes. They are now adapting these methods to discover the metabolites they produce. In collaboration with Anton Korobeynikov and Alexander Shlemov at Saint Petersburg State University, the researchers are planning to speed up the method and apply it for discovering novel antibiotics from metagenomes.
This research relies on computing infrastructure provided by the NIH Center for Computational Mass Spectrometry at UC San Diego. The Mass Spectrometry Innovation Center at UC San Diego, St. Petersburg State University and The University of Tokyo also took part in the research.
The work was funded partially by the National Institutes of Health and the Russian Science Foundation.
Dereplication of peptidic natural products through database search of mass spectra
Hosein Mohimani, Alexey Gurevich, Alla Mikheenko, Neha Garg, Louis-Felix Nothias, Akihiro Ninomiya, Kentaro Takada, Pieter C Dorrestein & Pavel A Pevzner*.
Share This:




Stay in the Know
Keep up with all the latest from UC San Diego. Subscribe to the newsletter today.